
![]()
This repository contains all Request for Comments (or rfcs) for the St. Jude Cloud project. Currently, RFCs on the St. Jude Cloud project are focused on outlining changes to the genomic analysis pipelines. However, we may expand to other areas over time.
🏠 Homepage
Process and Evaluation
Initiation of an RFC for the St. Jude Cloud project is currently limited to members of the St. Jude Cloud team. If you are a member of our community and have an idea you would like us to consider (or if you would like to propose your own RFC), please contact us before investing a large amount of time in your proposal.
Overview
All RFCs go through a three-stage process from draft to adoption:
- An initial draft is constructed by the author and discussed internally with members of the core St. Jude Cloud team.
- Titles of pull requests in this phase are typical prefixed with
[WIP]and should not be considered mature enough to accept comments from the community. - The amount of time the RFC spends in this state is variable depending on the scope of the proposal.
- Titles of pull requests in this phase are typical prefixed with
- The RFC opens for discussion from users and stakeholders1 within the St. Jude community.
- The beginning of this phase will be indicated by a comment on the pull request from the author.
- The RFC will remain in this phase for 1 week.
- The RFC opens for discussion from the broader community.
- The beginning of this phase will be indicated by a comment on the pull request from the author.
- The RFC will remain in this phase for 2 weeks.
Each phase of the process brings about further refinement of details and hardening of the proposal.
1 St. Jude community stakeholders include the Department of Computational Biology, Center for Applied Bioinformatics, and users of St. Jude Cloud that have requested data from a @stjude.org e-mail address.
Evaluation and acceptance
RFCs must have majority support within the St. Jude Cloud team before being adopted. In practice, most RFCs already have a significant amount of internal support before being drafted, so the rejection of an RFC will be exceedingly rare. The intent of these discussions is mostly geared towards gaining feedback from the community.
We welcome all ideas and concerns as we seek to understand how the needs of the genomics community evolve over time. Our ultimate goal is to make informed decisions that will benefit the majority of our users. That said, the project's direction is not driven strictly by community consensus and ultimately will be decided by the St. Jude Cloud team based on our best judgement.
Creating a proposal
- Create an initial draft of the RFC by creating a pull request on this repo.
- Create a branch on this repo with the form
<username>/<featurename>. - On that branch, copy the
0000-template.mdfile to thetext/folder and replace "template" with an appropriate feature name (e.g.text/rnaseq-workflow-v2). - If necessary, you can create a folder at
resources/<filename.md>/to hold any images or supporting materials (e.g.resources/0000-rnaseq-workflow-v2.0/) - Ensure that your pull request has
[WIP]prefixing the title as long as it is not ready for review!
- Create a branch on this repo with the form
- Open up the discussion for the community.
- Ensure your pull request's main comment has a "rendered" tag for easy review (see this example).
- Remove the
[WIP]if necessary. - Assign yourself as the assignee and add any relevant community members you'd like to review.
- Once the RFC has reached a consensus, it is ready for inclusion in the main repository.
- An owner of this repository will comment on the pull request with an assigned RFC number.
- Change your filename and resources folder to reflect this change (e.g. rename
text/0000-rnaseq-workflow-v2.0.mdtotext/1234-rnaseq-workflow-v2.0.md). - Ensure all of your links still work in your rendered copy.
- Merge in the PR and delete the branch.
Install
cargo install mdbook
Usage
mdbook build
python3 -m http.server -d book
# visit the rendered version in your browser at http://localhost:8000.
Author
👤 St. Jude Cloud Team
- Website: https://stjude.cloud
- Github: @stjudecloud
- Twitter: @StJudeResearch
Contributing
Contributions, issues and feature requests are welcome!
Feel free to check issues page. You can also take a look at the contributing guide.
📝 License
Copyright © 2020 St. Jude Cloud Team.
This project is MIT licensed.
Questions
With any questions, please contact us.
RNA-Seq v2 Pipeline
Introduction
This RFC lays out the specification for the RNA-Seq mapping pipeline v2.0.0. The improvements contained within are largely based on (a) new version of tools/reference files and (b) feedback from the community. You can find the relevant discussion on the associated pull request.
Motivation
- Tool additions and updates. The tools used in the first version of the RNA-seq pipeline are now woefully out of date (~2 years old). We desire to upgrade the tools to their latest published versions available so we can enjoy the benefits they provide. Furthermore, we'd like to explore adding many more tools to begin publishing QC results (probably a different QC pipeline in the future). See the section below on tool additions and updates for more details.
- Updated reference files. No changes have really been made to the
GRCh38_no_altanalysis set reference genome. However, three major releases of the GENCODE gene model have transpired since we released the first revision of the RNA-Seq workflow. GENCODE v31 is the latest at the time of writing, so we'd like to utilize the new information contained there as well. - Quality of life improvements based on feedback from the community. Input from the community has greatly informed our thoughts on what can be improved in this release. Some ideas that come to mind are:
- Removal of the
ERCC SpikeInsequences to ensure consistent sequence dictionaries between DNA and RNA data.- Popular tools such as
GATKandpicardare generally unhappy if the sequence dictionaries don't match perfectly. - The inclusion of the External RNA Controls Consortium (ERCC) Spike-in Control RNA sequences in the alignment reference file we used for RNA-Seq mapping was hence causing issues when using mapped RNA-Seq BAM files in conjunction with other non-RNA-Seq BAM files in downstream analysis using these tools.
- Last, many of our RNA-Seq samples were not generated using 'ERCC' spike-in control sequences, so the benefit its inclusion provides is minimal.
- Popular tools such as
- Removal of the
Discussion
Tool additions and upgrades
As part of the RNA-Seq workflow v2.0.0, multiple tools will be added and upgraded:
fq v0.2.0(Released November 28, 2018) will be added. This tool will be used to validate the output ofpicard SamToFastq.picard SamToFastqdoes not currently catch all of the errors we wish to catch at this stage (such as duplicate read names in the FastQ file). Thus, we will leverage this tool to independently validate that the data is well-formed by our definition of that phrase.ngsderive v1.0.2(Released March 3, 2020) will be added. This tool will be used to empirically infer strandedness of RNA-seq experiments.- Update
STAR 2.5.3a(Released March 17, 2017) toSTAR 2.7.1a(Released May 15, 2019). Upgraded to receive the benefits of bug fixes and software optimizations. - Update
samtools 1.4.0(Released March 13, 2017) tosamtools 1.9(Released July 18, 2018). Updating the samtools version whenever possible is of particular interest to me due to the historical fragility of the samtools code (although it has seemed to get better over the last year or so). - Update
picard 2.9.4(Released June 15, 2017) topicard 2.20.2(Released May 28, 2019). Upgraded to receive the benefits of bug fixes and software optimizations.
GENCODE compatability
One of the major discussions during this round of revisions was the compatability of the GRCh38_no_alt reference genome with the latest GENCODE gene set. It was posed as the following question:
This has just been an outstanding question of mine for a while — how big of an impact (if any at all) does the mismatch between the patch builds have? GENCODE v31 is built against
GRCh38.p12(see the in the title on the webpage), but obviously the no alt analysis set is derived fromGRCh38.p0(with the pseudoautosomal regions hard masked, the EBV chromosome added, etc.)
As is apparent in the question, the GRCh38 reference genome is regularly patched with non-coordinate altering, backwards compatable changes (see more information here). On the other hand, each GENCODE gene model release is based on a particular patch of the reference genome. This may be problematic because most bioinformatics analyses use a reference genome based on the non-patched version of GRCh38. What follows is our discussion and investigation into the effect between mismatching nucleotide sequences in the reference genome and the gene model.
First, we researched what some of the projects we respect in the community are doing:
| Pipeline | Reference Genome | Reference Genome Patch | Gene Model | Gene Model Patch |
|---|---|---|---|---|
| GDC's mRNA-Seq pipeline | GRCh38_no_alt-based w/ decoys + viral | GRCh38.p0 | GENCODE v22 | GRCh38.p2 |
| ENCODE's RNA-Seq pipeline | GRCh38_no_alt-based w/ SpikeIns | GRCh38.p0 | GENCODE v24 | GRCh38.p5 |
| Broad Institute's GTEx + TOPMed RNA-Seq pipeline | Broad's GRCh38 w/ ERCC SpikeIn | GRCh38.p0 | GENCODE v26 | GRCh38.p10 |
Note: You can confirm which patch the GENCODE genesets is based on just by clicking on the hyperlink. Verifying that each of these reference genomes is really based on GRCh38_no_alt takes a little bit more elbow grease: if you're interested, you can check out the comparison table in the appendix. If you are really interested, you can recapitulate those results by running the associated Jupyter notebook.
Based on the results of the above investigation, I reached out to the author of STAR, Alex Dobin, to get his opinion on whether the differences might affect some results. You can read my question and his reply here. In short, he confirms that, yes, this may alter results for the various analysis types we were interested in.
Given the landscape of the community and the author's response, we considered three possible options for moving ahead:
- We could try using the reference FASTA supplied with the respective GENCODE release as suggested by Dr. Dobin. This was the most undesirable approach from our groups perspective:
- The main reason was that this reference genome did not mirror the sequence dictionary or characteristics of the reference genome we use in our DNA-Seq pipeline out of the box. As outlined in the motivation section of this document, this incongruency was a major driving factor for the creation of this RFC.
- We agreed it would require too large an amount of postprocessing of the GENCODE reference genome to convert to apply all of the no alt analysis set changes (e.g. converting to UCSC names, masking regions of the genome, inserting the EBV chromosome).
- Additionally, this could leave room for the introduction of lots of strange errors, and there was no interest in getting into the business of genome generation (there is a reason no one applies the patches to their no alt analysis set).
- We could downgrade the GENCODE gene model to the latest release that was still based on
GRCh38.p0(GENCODE v21 would the correct version to use in this case).- The concordance of the reference sequences obviously made this choice an attractive option. However,
GENCODE v21was released over 5 years ago (06.2014) and there were many valuable updates over that time. In particular, a quick look showed that there were many more transcripts and that the number of lncRNAs more than doubled (see appendix). We did not want to lose all of this forward progress if we could avoid it. To see what we looked at, you can see the relevant section in the appendix.
- The concordance of the reference sequences obviously made this choice an attractive option. However,
- We could use the latest GENCODE release despite the mismatches outlined above as it appears most other projects have done.
- The general consensus was that the effect of this discordance would be small enough to tolerate so that we could gain all of the knowledge accumulated since the older release.
- To quantify this, we measured the differences in gene expression (as measured by the
R^2value betweenGENCODE v21andGENCODE v31) and splice junction detection (as measured by the number of splice junctions detected and the relative proportions of novel/partially novel/known junctions).
After discussion with the group, we decided to stick with option #3.
Gene model post-processing
Originally, I had posed this question to the group:
- Previously, we were filtering out anything not matching "level 1" or "level 2" from the gene model. This was due to best practices outlined during our RNA-Seq Workflow v1.0 discussions. I propose we revert this for the following reasons:
- The first sentence in section 2.2.2 of the STAR 2.7.1.a manual: "The use of the most comprehensive annotations for a given species is strongly recommended". So it seems the author recommends you use the most comprehensive gene model.
- Here is what the GENCODE FAQ has to say about the level 3 annotations: "Ensembl loci where they are different from the Havana annotation or where no Havana annotation can be found". Given that the GENCODE geneset is the union of automated annotations from the
Ensembl-genebuildand manual curation of theEnsembl-Havanateam, this level should be harmless in the event that levels 1 & 2 don't apply.- Last, the various other pipelines in the community don't tend to remove these features:
- The GDC documentation does not currently describe any filtering of their GENCODE v22 GTF (see the section labeled "Step 1: Building the STAR index").
- ENCODE is not filtering any features, they are just changing some of the contig names in the GENCODE v24 GTF (see the analysis in the appendix).
- The TOPMed pipeline does outline some postprocessing they are doing to the GENCODE v26 GTF, but it's mostly just collapsing exons. I inspected the script, which does have some functionality built in to blacklist a list of transcript IDs. However, the documentation does not specify that this is turned on by default.
After discussion internally, we decided to discontinue removing level 3 annotations by default. This is more consistent with what is being done in the community and it was decided that this was the most straightforward method with little associated risk. Therefore we are no longer performing any post-processing of the gene model.
Quality of life improvements
- Add
picard ValidateSamFileto the checks after theSTARalignment andpicard SortSamsteps. The criticism internally is thatValidateSamFileis quite stringent and often errors with concerns we don't care about. I'm testing this out as I develop the pipeline, and so far, I've found the following warnings to be ignore-worthy:INVALID_PLATFORM_VALUEis pretty annoying. It just complains if a read group doesn't contain aPLattribute. I'm not sure it's worth going back and fixing these.MISSING_PLATFORM_VALUE. Similar toINVALID_PLATFORM_VALUE, some of our samples have read groups with missing platform values and so we ignore those errors for now.
- For dependency management, we have moved to using
condawithin standard docker images. All packages should be available within thedefaults,conda-forge, andbiocondarepositories. - Add a checksum algorithm and publish the results in the data browser. After consideration internally, we decided to use the
md5sumchecksum because of its ubiquity.
Various other changes
- Removed a section of the pipeline that reformatted the header of the BAM to be cleaner.
STARoutputs a header that is formatted fine already, and I found this code to just be an area where an error could be introduced for little benefit. - Removed a section of custom code that checks for duplicate read groups.
picard ValidateSamFiledoes this for you (see the documentation for this tool. Specifically, theDUPLICATE_READ_GROUP_IDerror).
Specification
Dependencies
If you'd like the full conda environment, you can install it using the following command. Obviously, you'll need to install anaconda first.
conda create -n star-mapping \
-c conda-forge \
-c bioconda \
picard==2.20.2 \
samtools==1.9 \
star==2.7.1a \
htseq==0.11.2 \
ngsderive==1.0.2 \
-y
Additionally, you will want to install our fqlib library to check that FastQ files are properly paired and have no duplicate read names. Installation of the Rust programming language is required.
cargo install --git https://github.com/stjude/fqlib.git --tag v0.3.1
Reference files
The following reference files are used as the basis of the RNA-Seq Workflow v2.0.0:
-
Similarly to all analysis pipelines in St. Jude Cloud, we use the
GRCh38_no_altanalysis set for our reference genome. You can get a copy of the file here. Additionally, you can get the file by running the following commands:wget ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz -O GRCh38_no_alt.fa.gz gunzip GRCh38_no_alt.fa.gz echo "a6da8681616c05eb542f1d91606a7b2f GRCh38_no_alt.fa" > GRCh38_no_alt.fa.md5 md5sum -c GRCh38_no_alt.fa.md5 # > GRCh38_no_alt.fa: OK -
For the gene model, we use the GENCODE v31 "comprehensive gene annotation" GTF for the "CHR" regions. You can get a copy of the gene annotation file here. For the exact steps to generate the gene model we use, you can run the following commands:
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_31/gencode.v31.annotation.gtf.gz echo "0aac86e8a6c9e5fa5afe2a286325da81 gencode.v31.annotation.gtf.gz" > gencode.v31.annotation.gtf.gz.md5 md5sum -c gencode.v31.annotation.gtf.gz.md5 # > gencode.v31.annotation.gtf.gz: OK gunzip gencode.v31.annotation.gtf.gz echo "4e22351ae216e72aa57cd6d6011960f8 gencode.v31.annotation.gtf" > gencode.v31.annotation.gtf.md5 md5sum -c gencode.v31.annotation.gtf.md5 # > gencode.v31.annotation.gtf: OK -
Last, the following command is used to prepare the STAR index file:
STAR --runMode genomeGenerate \ # Use genome generation runMode. --genomeDir $OUTPUT_DIR \ # Specify an output directory. --runThreadN $NCPU \ # Number of threads to use to build genome database. --genomeFastaFiles $FASTA \ # A path to the GRCh38_no_alt.fa FASTA file. --sjdbGTFfile $GENCODE_GTF_V31 \ # GENCODE v31 gene model file. --sjdbOverhang 125 # Splice junction database overhang parameter, the optimal value is (Max length of RNA-Seq read-1).
Workflow
Here are the resulting steps in the RNA-Seq Workflow v2.0.0 pipeline. There might be slight alterations in the actual implementation, which can be found in the St. Jude Cloud workflows repository.
-
Run
picard ValidateSamon the incoming BAM to ensure that it is well-formed enough to convert back to FastQ.picard ValidateSamFile I=$INPUT_BAM \ # Input BAM. IGNORE=INVALID_PLATFORM_VALUE \ # Validations to ignore. IGNORE=MISSING_PLATFORM_VALUE -
Split BAM file into multiple BAMs on the different read groups using
samtools split. See the samtools documentation for more information.samtools split -u $UNACCOUNTED_BAM_NAME \ # Reads that do not belong to a read group or the read group is unrecognized go here. -f '%*_%!.%.' # Format of output BAM file names.If the BAM has unaccounted reads, those will need to be triaged and this step will need to be rerun.
-
Run Picard
SamToFastqon each of the BAMs generated in the previous step.picard SamToFastq \ INPUT=$INPUT_BAM \ FASTQ=$FASTQ_R1 \ SECOND_END_FASTQ=$FASTQ_R2 \ RE_REVERSE=true \ VALIDATION_STRINGENCY=SILENT -
Run
fq linton each of the FastQ pairs that was generated in the previous step as a sanity check. You can see the checks that thefqtool performs here.fq lint $FASTQ_R1 $FASTQ_R2 # Files for read 1 and read 2. -
Run the
STARalignment algorithm.STAR --readFilesIn $ALL_FASTQ_R1 $ALL_FASTQ_READ2 \ # FastQ files, separated by comma if there are multiple. The order of your R1 and R2 files has to match! --outSAMattrRGline $ALL_RG \ # Read group lines in the same order as `readFilesIn` (derived from earlier `samtools split` step). --genomeDir $STARDB \ # Directory containing the STAR genome --runThreadN $NCPU \ # Number of threads to use. You must request the correct amount from HPCF first! --outSAMunmapped Within \ # Keep unmapped reads in the final BAM file. --outSAMstrandField intronMotif \ # Preserve compatibility with Cufflinks by including the XS attribute (strand derived from intron motif). --outSAMattributes NH HI AS nM NM MD XS \ # Recommended SAM attributes to include for compatibility. Refer to manual for specifics. --outFilterMultimapScoreRange 1 \ # Ensures that all multi-mapped reads will need to share the mapping score. --outFilterMultimapNmax 20 \ # Max number of multi-mapped SAM entries for each read. --outFilterMismatchNmax 10 \ # Max number of mismatches allowed from alignment. --alignIntronMax 500000 \ # Max intron size considered. --alignMatesGapMax 1000000 \ # Max gap allowed between two mates. --sjdbScore 2 \ # Additional weight given to alignments that cross database junctions when scoring alignments. --alignSJDBoverhangMin 1 \ # Minimum overhang required on each side of a splice junction. Here, we require only one base on each side. --outFilterMatchNminOverLread 0.66 \ # 66% of the read must be perfectly matched to the reference sequence. --outFilterScoreMinOverLread 0.66 \ # Score must be greater than 66% of the read length. So for RL=100, the alignment must have a score > 66. --limitBAMsortRAM $RAM_LIMIT \ # Amount of RAM to use for sorting. Recommended value is [Max amount of RAM] - 5GB. --outFileNamePrefix $OUT_FILE_PREFIX \ # All output files will have this path prepended. --twopassMode Basic # Use STAR two-pass mapping technique (refer to manual). -
Run
picard SortSamon theSTAR-aligned BAM file. Note that this is much more memory efficient than usingSTAR's built-in sorting (which often takes 100GB+ of RAM).picard SortSam I=$STAR_BAM \ # Input BAM. O=$SORTED_BAM \ # Coordinate-sorted BAM. SO="coordinate" \ # Specify the output should be coordinate-sorted CREATE_INDEX=false \ # Explicitly do not create an index at this step, in case the default changes. CREATE_MD5_FILE=false \ # Explicity do not create an md5 checksum at this step, in case the default changes. COMPRESSION_LEVEL=5 \ # Explicitly set the compression level to 5, although, at the time of writing, this is the default. VALIDATION_STRINGENCY=SILENT # Turn of validation stringency for this step. -
Index the coordinate-sorted BAM file.
samtools index $STAR_SORTED_BAM # STAR-aligned, coordinate-sorted BAM. -
Run
picard ValidateSamFileon the aligned and sorted BAM file.picard ValidateSamFile I=$STAR_SORTED_BAM \ # STAR-aligned, coordinate-sorted BAM. IGNORE=INVALID_PLATFORM_VALUE \ # Validations to ignore. IGNORE=MISSING_PLATFORM_VALUE -
Run
ngsderive'sstrandednesssubcommand to confirm that the lab's information on strandedness reflects what was is computed. Manually triage any discrepancies. This is particularly useful for historical samples. Additionally, if the value for strandedness isn't known at run time, we can use the inferred value (if reported).ngsderive strandedness $STAR_SORTED_BAM \ # STAR-aligned, coordinate-sorted BAM. -g $GENCODE_GTF_V31_GZ # GENCODE v31 GTF (gzipped) -
Next,
htseq-countis run for the final counts file to be delivered.htseq-count -f bam \ # Specify input file as BAM. -r pos \ # Specify the BAM is position sorted. -s $PROVIDED_OR_INFERRED \ # Strandedness as specified by the lab and confirmed by `ngsderive strandedness` above. Typically `reverse` for St. Jude Cloud data. -m union \ # For reference, GDC uses "intersection-nonempty". -i gene_name \ # I'd like to use the colloquial gene name here. For reference, GDC uses "gene_id" here. Needs input from reviewers. --secondary-alignments ignore \ # Elect to ignore secondary alignments. Needs input from reviewers. --supplementary-alignments ignore \ # Elect to ignore supplementary alignments. Needs input from reviewers. $STAR_SORTED_BAM \ # STAR-aligned, coordinate-sorted BAM. $GENCODE_GTF_V31 # GENCODE v31 GTF
Appendix
Reference genome comparison
Below are the results of an analysis of each pipeline's GRCh38-based reference genome. The steps are essentially:
- Research what reference genome each project is using and where to find it.
- Download it.
- Use
picard CreateSequenceDictionaryto get the md5sums for each sequence in the dictionary. - Compare for the common chromosomes in each reference (the autosomes, the sex chromosomes, and the EBV decoy).
If you are interested in seeing the full comparison table or in regenerating these results, you can see the associated Jupyter notebook.
| Sequence Name | NCBI (baseline) | ENCODE | GDC | TOPMed | Concordant |
|---|---|---|---|---|---|
| chr1 | 6aef897c3d6ff0c78aff06ac189178dd | 6aef897c3d6ff0c78aff06ac189178dd | 6aef897c3d6ff0c78aff06ac189178dd | 6aef897c3d6ff0c78aff06ac189178dd | True |
| chr2 | f98db672eb0993dcfdabafe2a882905c | f98db672eb0993dcfdabafe2a882905c | f98db672eb0993dcfdabafe2a882905c | f98db672eb0993dcfdabafe2a882905c | True |
| chr3 | 76635a41ea913a405ded820447d067b0 | 76635a41ea913a405ded820447d067b0 | 76635a41ea913a405ded820447d067b0 | 76635a41ea913a405ded820447d067b0 | True |
| chr4 | 3210fecf1eb92d5489da4346b3fddc6e | 3210fecf1eb92d5489da4346b3fddc6e | 3210fecf1eb92d5489da4346b3fddc6e | 3210fecf1eb92d5489da4346b3fddc6e | True |
| chr5 | a811b3dc9fe66af729dc0dddf7fa4f13 | a811b3dc9fe66af729dc0dddf7fa4f13 | a811b3dc9fe66af729dc0dddf7fa4f13 | a811b3dc9fe66af729dc0dddf7fa4f13 | True |
| chr6 | 5691468a67c7e7a7b5f2a3a683792c29 | 5691468a67c7e7a7b5f2a3a683792c29 | 5691468a67c7e7a7b5f2a3a683792c29 | 5691468a67c7e7a7b5f2a3a683792c29 | True |
| chr7 | cc044cc2256a1141212660fb07b6171e | cc044cc2256a1141212660fb07b6171e | cc044cc2256a1141212660fb07b6171e | cc044cc2256a1141212660fb07b6171e | True |
| chr8 | c67955b5f7815a9a1edfaa15893d3616 | c67955b5f7815a9a1edfaa15893d3616 | c67955b5f7815a9a1edfaa15893d3616 | c67955b5f7815a9a1edfaa15893d3616 | True |
| chr9 | 6c198acf68b5af7b9d676dfdd531b5de | 6c198acf68b5af7b9d676dfdd531b5de | 6c198acf68b5af7b9d676dfdd531b5de | 6c198acf68b5af7b9d676dfdd531b5de | True |
| chr10 | c0eeee7acfdaf31b770a509bdaa6e51a | c0eeee7acfdaf31b770a509bdaa6e51a | c0eeee7acfdaf31b770a509bdaa6e51a | c0eeee7acfdaf31b770a509bdaa6e51a | True |
| chr11 | 1511375dc2dd1b633af8cf439ae90cec | 1511375dc2dd1b633af8cf439ae90cec | 1511375dc2dd1b633af8cf439ae90cec | 1511375dc2dd1b633af8cf439ae90cec | True |
| chr12 | 96e414eace405d8c27a6d35ba19df56f | 96e414eace405d8c27a6d35ba19df56f | 96e414eace405d8c27a6d35ba19df56f | 96e414eace405d8c27a6d35ba19df56f | True |
| chr13 | a5437debe2ef9c9ef8f3ea2874ae1d82 | a5437debe2ef9c9ef8f3ea2874ae1d82 | a5437debe2ef9c9ef8f3ea2874ae1d82 | a5437debe2ef9c9ef8f3ea2874ae1d82 | True |
| chr14 | e0f0eecc3bcab6178c62b6211565c807 | e0f0eecc3bcab6178c62b6211565c807 | e0f0eecc3bcab6178c62b6211565c807 | e0f0eecc3bcab6178c62b6211565c807 | True |
| chr15 | f036bd11158407596ca6bf3581454706 | f036bd11158407596ca6bf3581454706 | f036bd11158407596ca6bf3581454706 | f036bd11158407596ca6bf3581454706 | True |
| chr16 | db2d37c8b7d019caaf2dd64ba3a6f33a | db2d37c8b7d019caaf2dd64ba3a6f33a | db2d37c8b7d019caaf2dd64ba3a6f33a | db2d37c8b7d019caaf2dd64ba3a6f33a | True |
| chr17 | f9a0fb01553adb183568e3eb9d8626db | f9a0fb01553adb183568e3eb9d8626db | f9a0fb01553adb183568e3eb9d8626db | f9a0fb01553adb183568e3eb9d8626db | True |
| chr18 | 11eeaa801f6b0e2e36a1138616b8ee9a | 11eeaa801f6b0e2e36a1138616b8ee9a | 11eeaa801f6b0e2e36a1138616b8ee9a | 11eeaa801f6b0e2e36a1138616b8ee9a | True |
| chr19 | 85f9f4fc152c58cb7913c06d6b98573a | 85f9f4fc152c58cb7913c06d6b98573a | 85f9f4fc152c58cb7913c06d6b98573a | 85f9f4fc152c58cb7913c06d6b98573a | True |
| chr20 | b18e6c531b0bd70e949a7fc20859cb01 | b18e6c531b0bd70e949a7fc20859cb01 | b18e6c531b0bd70e949a7fc20859cb01 | b18e6c531b0bd70e949a7fc20859cb01 | True |
| chr21 | 974dc7aec0b755b19f031418fdedf293 | 974dc7aec0b755b19f031418fdedf293 | 974dc7aec0b755b19f031418fdedf293 | 974dc7aec0b755b19f031418fdedf293 | True |
| chr22 | ac37ec46683600f808cdd41eac1d55cd | ac37ec46683600f808cdd41eac1d55cd | ac37ec46683600f808cdd41eac1d55cd | ac37ec46683600f808cdd41eac1d55cd | True |
| chrX | 2b3a55ff7f58eb308420c8a9b11cac50 | 2b3a55ff7f58eb308420c8a9b11cac50 | 2b3a55ff7f58eb308420c8a9b11cac50 | 2b3a55ff7f58eb308420c8a9b11cac50 | True |
| chrY | ce3e31103314a704255f3cd90369ecce | ce3e31103314a704255f3cd90369ecce | ce3e31103314a704255f3cd90369ecce | ce3e31103314a704255f3cd90369ecce | True |
| chrM | c68f52674c9fb33aef52dcf399755519 | c68f52674c9fb33aef52dcf399755519 | c68f52674c9fb33aef52dcf399755519 | c68f52674c9fb33aef52dcf399755519 | True |
| chrEBV | 6743bd63b3ff2b5b8985d8933c53290a | 6743bd63b3ff2b5b8985d8933c53290a | 6743bd63b3ff2b5b8985d8933c53290a | 6743bd63b3ff2b5b8985d8933c53290a | True |
ENCODE GTF generation
A small investigation was done to see how ENCODE's GENCODE v24 GTF was being produced (the md5sum between the original GENCODE v24 primary assembly file and ENCODE's version of the file weren't matching up). In short, they just changed the names of some of the contigs. You can verify this yourself by doing an md5sum without the sequence name column of the GTF (or manually download and inspect the differences like I did :)).
curl -sL ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_24/gencode.v24.primary_assembly.annotation.gtf.gz | \
gunzip -c | \
cut -f2- -d$'\t' | \
md5sum
# 5227fac91c8d32b3fa3a8f78c4bf0e5c -
curl -sL https://www.encodeproject.org/files/gencode.v24.primary_assembly.annotation/@@download/gencode.v24.primary_assembly.annotation.gtf.gz | \
gunzip -c | \
cut -f2- -d$'\t' | \
md5sum
# 5227fac91c8d32b3fa3a8f78c4bf0e5c -
GENCODE feature comparisons
Below are a few commands used to quickly evaluate how much the GENCODE geneset has changed over time. This was useful in our discussion about how much value would be lost if we just used GENCODE v21 (which was based on GRCh38.p0). See the discussion on the GENCODE compatibility for more information.
-
Commands that were used in the comparison of feature types between
GENCODE v21andGENCODE v31:gawk '$0 !~ /^#/ { features[$3] += 1; total += 1 } END {for (key in features) { print "Feature " key ": " features[key] " (" (features[key]/total)*100 "%)" }; print "Total: " total}' gencode.v21.annotation.gtf # Feature exon: 1162114 (45.6341%) # Feature CDS: 696929 (27.3671%) # Feature UTR: 275100 (10.8027%) # Feature gene: 60155 (2.36217%) # Feature start_codon: 81862 (3.21457%) # Feature Selenocysteine: 114 (0.00447657%) # Feature stop_codon: 73993 (2.90557%) # Feature transcript: 196327 (7.7094%) # Total: 2546594 gawk '$0 !~ /^#/ { features[$3] += 1; total += 1 } END {for (key in features) { print "Feature " key ": " features[key] " (" (features[key]/total)*100 "%)" }; print "Total: " total}' gencode.v31.annotation.gtf # Feature exon: 1363843 (47.3247%) # Feature CDS: 755320 (26.2092%) # Feature UTR: 308315 (10.6984%) # Feature gene: 60603 (2.10289%) # Feature start_codon: 87299 (3.02923%) # Feature Selenocysteine: 119 (0.00412924%) # Feature stop_codon: 79505 (2.75878%) # Feature transcript: 226882 (7.87269%) # Total: 2881886 -
Commands that were used in the comparison of
gene_typesforgenefeatures betweenGENCODE v21andGENCODE v31:gawk '$3 ~ /gene/' gencode.v21.annotation.gtf | gawk 'match($0, /gene_type "([A-Za-z0-9]+)"/, a) { types[a[1]] += 1; total += 1 } END {for (key in types) { print "Gene type " key ": " types[key] " (" (types[key]/total)*100 "%)" }; print "Total: " total}' # Gene type rRNA: 549 (2.54508%) # Gene type antisense: 5542 (25.6919%) # Gene type pseudogene: 29 (0.13444%) # Gene type lincRNA: 7666 (35.5385%) # Gene type TEC: 1058 (4.90473%) # Gene type miRNA: 3837 (17.7878%) # Gene type snoRNA: 978 (4.53386%) # Gene type snRNA: 1912 (8.86375%) # Total: 21571 gawk '$3 ~ /gene/' gencode.v31.annotation.gtf | gawk 'match($0, /gene_type "([A-Za-z0-9]+)"/, a) { types[a[1]] += 1; total += 1 } END {for (key in types) { print "Gene type " key ": " types[key] " (" (types[key]/total)*100 "%)" }; print "Total: " total}' # Gene type ribozyme: 8 (0.0351463%) # Gene type scaRNA: 49 (0.215271%) # Gene type lncRNA: 16840 (73.983%) # Gene type rRNA: 52 (0.228451%) # Gene type vaultRNA: 1 (0.00439329%) # Gene type scRNA: 1 (0.00439329%) # Gene type sRNA: 5 (0.0219664%) # Gene type pseudogene: 18 (0.0790792%) # Gene type TEC: 1064 (4.67446%) # Gene type miRNA: 1881 (8.26377%) # Gene type snoRNA: 942 (4.13848%) # Gene type snRNA: 1901 (8.35164%) # Total: 22762 -
The following is a quick command that can be used on a GENCODE GTF to summarize the level counts/percentages in the GTF file. This was used to quantify how much of the GTF would be eliminated when all level 3 features were removed.
gawk 'match($0, /level ([0-9]+)/, a) { levels[a[1]] += 1; total += 1 } END {for (key in levels) { print "Level " key ": " levels[key] " (" (levels[key]/total)*100 "%)" }; print "Total : " total}' gencode.v31.annotation.gtf # Level 1: 186461 (6.4701%) # Level 2: 2450972 (85.0475%) # Level 3: 244453 (8.4824%) # Total : 2881886
scRNA-Seq Pipeline
Introduction
This RFC lays out the specification for the scRNA-Seq mapping pipeline.
Motivation
To provide the community access to data from scRNA-Seq experiments performed at St. Jude Children's Research Hospital, we propose the following data harmonization pipeline. The goal of this pipeline is to provide harmonized alignments for scRNA-Seq data. For this pipeline, we will make no recommendations on downstream analysis, focusing instead on harmonizing the underlying sequencing data and leaving analysis decisions to the user.
Discussion
The development of bulk RNA-Seq enabled efficient profiling of transcripts in a sample. The ability to interrogate the transcriptome in an unbiased manner, largely replaced previous methods such as microarrays or RT-qPCR. A bulk RNA-Seq experiment sample is a mixture of cells. This method is useful for analyses such as comparing expression between tumor and normal samples. However, this limits the analysis to the average expression over a population of cells and is unable to capture the heterogeneity of gene expression across individual cells. Newer methods have enabled RNA-Seq to be applied to single cells. This enables new questions about cell-specific transcriptome changes. This methodology has been employed in large projects such as the Human Cell Atlas to capture the cellular diversity within organisms.
scRNA-Seq methodology
Generally, scRNA-Seq differs somewhat from bulk RNA-Seq. Most approaches generate three key pieces of information: 1. cDNA fragment from the RNA transcript, 2. a cell barcode that identifies in which cell the RNA was expressed, and 3. a unique molecular identifier (UMI) to enable collapse of PCR duplicates.
There are several commerical options for performing scRNA-Seq. One such option is the 10x Chromium platform. In their approach using paired-end reads, the celluar barcode and UMI are found in read 1 and the transcript sequence is found in read 2. The 10x method is the most popular commercially available method, so handling data for that platform will be the priority.
The canonical tool for processing 10x Chromium scRNA-Seq data is their Cell Ranger tool. This tool uses STAR to perform alignment. It then uses the transcriptome annotation GTF to label reads as exonic, intronic, or intergenic. It then performs some additional refinement on the alignments to determine which reads are transcriptomic. These reads are then carried into UMI counting. The UMI counting step produces a raw gene by cell matrix of counts.
Processing Pipeline
For processing of bulk RNA-Seq data, a common standard workflow is to align against the genome using STAR. Quantification is then done using a package such as HTSeq-count. An example of a bulk RNA-Seq pipeline is the St. Jude Cloud RNA-Seq standard workflow v2.0.0.
Aligner choice
For consistency with other St. Jude Cloud datasets, we are not considering psuedoalignment methods such as kallisto and Alevin. This leaves the previously described Cell Ranger method and STARsolo as the primary candidates for use in St. Jude Cloud.
Bruning, et. al. (2022) provide a useful comparison of commonly used scRNA-Seq methods. However due to our preference for an alignment-based method, we will choose between Cell Ranger and STARsolo. Cell Ranger is the most commonly used method for processing data generated from the 10x protocol. Downstream analysis tools have been developed to directly ingest data in the formats produced by Cell Ranger. The tool is an integrated system that performs alignment, feature counting, and quality control.
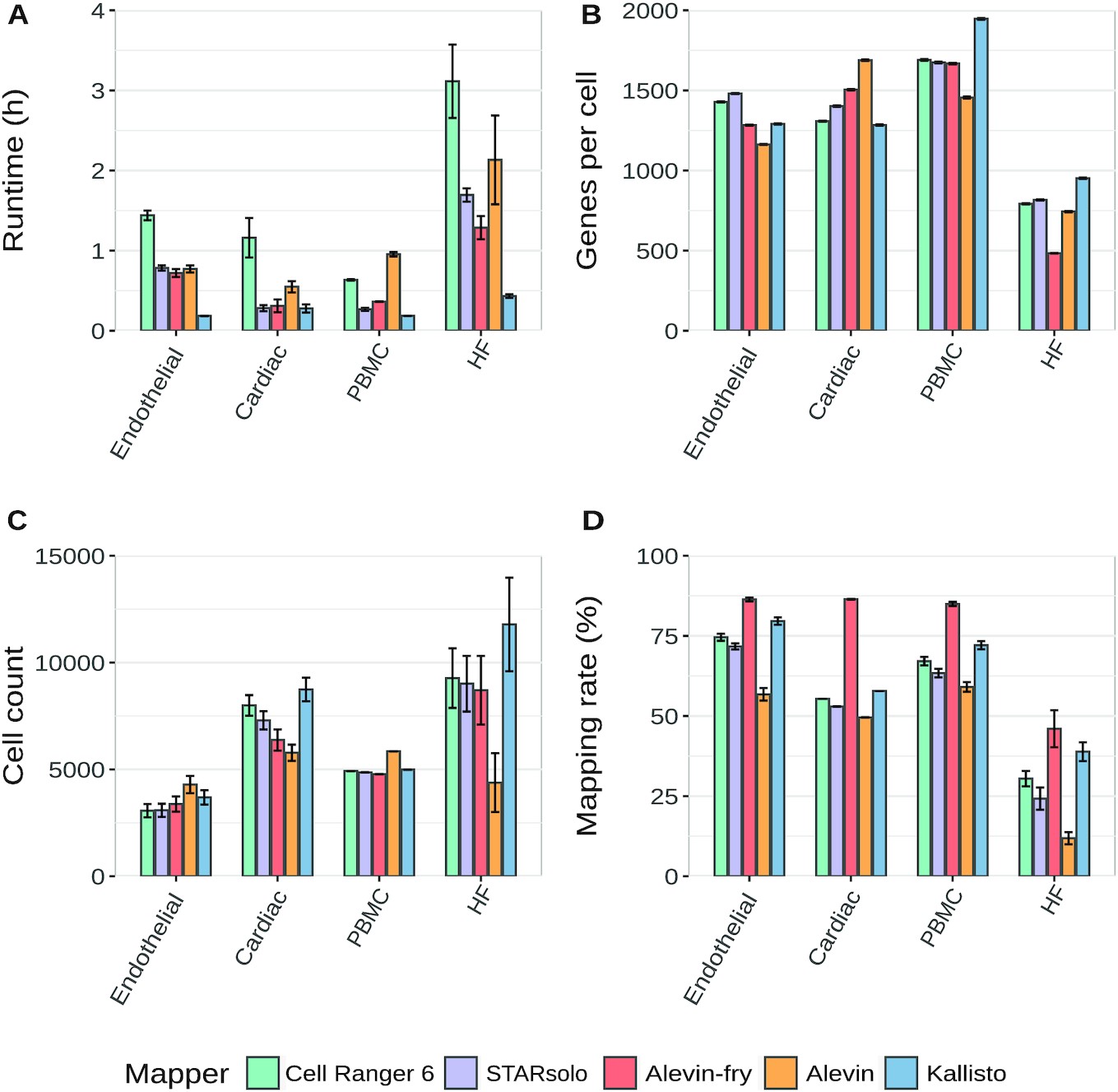
Quantification choice
Quantification is a product of both the Cell Ranger and STARsolo packages. Quantification could also be accomplished using a pseudoalignment method, such as Kallisto or Alevin, if there was no desire to share the raw data in BAM format. However that would be a departure from the normal St. Jude Cloud methodology, so that will not be discussed here. Therefore, there is no additional quantification choice to be made beyond that of the aligner package.
Using either STARsolo or Cell Ranger for analysis requires selection of reference files. For the genome, we will continue using GRCh38_no_alt. The transcript annotation GTF is more complicated. 10x provides several versions of the annotation files used in Cell Ranger. For simplicity, we will use the Cell Ranger reference files version 2020-A which incorporates GENCODE v32.
Specification
Metadata
Sample-based
- Library Prep Kit - e.g. Illumina TruSeq RNA Library Prep Kit V2
- Library Selection Protocol - e.g. Total, Poly-A
- Sequencing Machine - e.g. Illumina HiSeq 2000, Illumina NovaSeq 6000
- Library Layout - Single-End, Paired-End
- RNA-Seq Strandedness - Unstranded, Stranded-Reverse, Stranded-Forward
- Read Length - Number of bp per read
- Tissue Preservative - e.g. FFPE, Fresh/Frozen
- Cell Isolation Protocol - e.g. Droplet, Microfluidics
- Flowcell design - Multiplexed, split across flowcells, etc.
Cell-based
TODO
Dependencies
If you'd like the full conda environment, you can install it using the following command. Obviously, you'll need to install anaconda first.
conda create -n scrnaseq-mapping \
-c conda-forge \
-c bioconda \
picard==2.20.2 \
samtools==1.9 \
ngsderive==2.2.0 \
tabix==1.11 \
-y
Additionally, you will want to install our fqlib library to check that FastQ files are properly paired and have no duplicate read names. Installation of the Rust programming language is required.
cargo install --git https://github.com/stjude/fqlib.git --tag v0.8.0
Additionally, you will need to install Cell Ranger from 10x genomics to perform the alignment and feature calling.
curl -o cellranger-7.0.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-7.0.0.tar.gz?Expires=1659064381&Policy=eyJTdGF0ZW1lbnQiOlt7IlJlc291cmNlIjoiaHR0cHM6Ly9jZi4xMHhnZW5vbWljcy5jb20vcmVsZWFzZXMvY2VsbC1leHAvY2VsbHJhbmdlci03LjAuMC50YXIuZ3oiLCJDb25kaXRpb24iOnsiRGF0ZUxlc3NUaGFuIjp7IkFXUzpFcG9jaFRpbWUiOjE2NTkwNjQzODF9fX1dfQ__&Signature=SGKFTznrgspKBPQ2oRZ5C9KazslNWsTkxtkGnnaaPtrOR2iihhvkJ5frOlT5ahkABzBlHf~8EjLqcJ6HofbiQ4McbrHsmAnRggfv2QK0WScF40qDE3kGm~Z57VumO5homkdJPEf9r5DlbMzlArE5-UBH91F0rMMribGjwABXJCjUsAuet0klbUg~~SzCxoNMfTvo3Nn7Mxv7ls51LQf72riNit9zne6WsHynIY7YBHaquVftTi-C6bMZw5A3NoekyA7LBTZwc6sFjrsFrf3s3BpYKeRkBtPdkDMljME9JLDo9wXM7Lf1SfTj1vtNVUDoNhMnTFplDNzyBaDEDm7GVg__&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA"
echo '30855cb96a097c9cab6b02bdb520423f cellranger-7.0.0.tar.gz' > cellranger-7.0.0.tar.gz.md5
md5sum -c cellranger-7.0.0.tar.gz.md5
tar -xzvf cellranger-7.0.0.tar.gz
export PATH=$(pwd)/cellranger-7.0.0:$PATH
Reference files
The following reference files are used as the basis of the scRNA-Seq Workflow v1.0.0:
-
We will use the 10x provided reference files version 2020-A. You can get the file by running the following commands:
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz -O 10x-GRCh38-2020-A.tar.gz echo "dfd654de39bff23917471e7fcc7a00cd 10x-GRCh38-2020-A.tar.gz" > 10x-GRCh38-2020-A.tar.gz.md5 md5sum -c 10x-GRCh38-2020-A.tar.gz.md5 # > 10x-GRCh38-2020-A.tar.gz: OK tar -zxf 10x-GRCh38-2020-A.tar.gz mv refdata-gex-GRCh38-2020-A 10x-GRCh38-2020-A
Workflow
Here are the resulting steps in the scRNA-Seq Workflow v1.0.0 pipeline. There might be slight alterations in the actual implementation, which can be found in the St. Jude Cloud workflows repository.
-
Run
picard ValidateSamon the incoming BAM to ensure that it is well-formed enough to convert back to FastQ.picard ValidateSamFile \ I=$INPUT_BAM \ # Input BAM. IGNORE=INVALID_PLATFORM_VALUE \ # Validations to ignore. IGNORE=MISSING_PLATFORM_VALUE -
Split BAM file into multiple BAMs on the different read groups using
samtools split. See the samtools documentation for more information.samtools split -u $UNACCOUNTED_BAM_NAME \ # Reads that do not belong to a read group or the read group is unrecognized go here. -f '%*_%!.%.' # Format of output BAM file names.If the BAM has unaccounted reads, those will need to be triaged and this step will need to be rerun.
-
Run Picard
SamToFastqon each of the BAMs generated in the previous step.picard SamToFastq \ INPUT=$INPUT_BAM \ FASTQ=$FASTQ_R1 \ SECOND_END_FASTQ=$FASTQ_R2 \ RE_REVERSE=true \ VALIDATION_STRINGENCY=SILENT -
Run
fq linton each of the FastQ pairs that was generated in the previous step as a sanity check. You can see the checks that thefqtool performs here.fq lint $FASTQ_R1 $FASTQ_R2 # Files for read 1 and read 2. -
Run
Cell Ranger.cellranger count \ --id=$OUTPUT_DIR_NAME \ # A unique run id and output folder name [a-zA-Z0-9_-]+ --transcriptome=$REFERENCE \ # Path of folder containing 10x-compatible transcriptome reference --fastqs=$FASTQ_DIR \ # Path to input FASTQ data --sample=$FASTQ_PREFIX \ # Prefix of the filenames of FASTQs to select --localcores=$N_CORES \ # Maximum number of cores to use --localmem=$MEM_GB # Maximum memory (GB) to use -
Run
picard ValidateSamFileon the aligned and sorted BAM file.picard ValidateSamFile \ I=$CELL_RANGER_SORTED_BAM \ # Cell Ranger-aligned, coordinate-sorted BAM. IGNORE=INVALID_PLATFORM_VALUE \ # Validations to ignore. IGNORE=MISSING_PLATFORM_VALUE -
Run
ngsderive'sstrandednesssubcommand to confirm that the lab's information on strandedness reflects what was is computed. Manually triage any discrepancies. This is particularly useful for historical samples. Additionally, if the value for strandedness isn't known at run time, we can use the inferred value (if reported).ngsderive strandedness $CELL_RANGER_SORTED_BAM \ # Cell Ranger-aligned, coordinate-sorted BAM. -g $GENCODE_GTF_V32_GZ # GENCODE v32 GTF (gzipped)
Drafts
The following are candidate RFCs that are being rendered for easy review. They are not accepted St. Jude Cloud RFCs. For more information please see the associated pull request.